 HOME | ÍNDICE POR TÍTULO | NORMAS PUBLICACIÓN
HOME | ÍNDICE POR TÍTULO | NORMAS PUBLICACIÓN Espacios. Vol. 37 (Nº 31) Año 2016. Pág. 6
Antonio Pedro BRUSAMARELLO 1; Paulo Henrique de OLIVEIRA; Taciane FINATTO; Michelangelo Muzell TREZZI; Micheli NEGRI; Cheila da Silva CHAGAS
Recibido: 03/06/16 • Aprobado: 03/07/2016
ABSTRACT: The isolation of DNA (deoxyribonucleic acid) of weeds is a basic knowledge of fundamental importance to the advancement in molecular genetic studies, such molecular markers, DNA sequencing and identification of mutations. In this study, we determined the best low-cost method for genomic DNA isolation from Euphorbia heterophylla L. The experimental design was completely randomized with four replications. We analyzed 10 methods for DNA isolation, which consisted in adaptations of methods, described in the literature. We have measured the amount of DNA isolated, its purity, integrity and interference in the PCR (polymerase chain reaction). The 0.2%BME method is the most suitable for DNA isolation of E. heterophylla, because it does not cause contamination with polyphenols, ensures high quantity and quality of isolated DNA and demanding less time to the execution of isolation steps. |
RESUMO: O isolamento de DNA (ácido desoxirribonucleico) de espécies daninhas é conhecimento básico de fundamental importância para o avanço nos estudos de genética molecular, como o desenvolvimento de marcadores moleculares, sequenciamento de DNA e identificação de mutações. O presente trabalho teve por objetivo determinar o melhor método de baixo custo para isolamento de DNA genômico de Euphorbia heterophylla L. O delineamento experimental foi inteiramente casualizado com quatro repetições. Foram testados 10 métodos de isolamento de DNA que consistiram em adaptações de métodos descritos na literatura. Foram avaliadas a quantidade de DNA isolado, sua pureza, integridade e interferência na PCR (reação em cadeia da polimerase). O método 0.2%BME é o mais indicado para o isolamento de DNA da espécie E. heterophylla, pois não ocasiona contaminação por polifenóis, garante alta quantidade e qualidade do DNA isolado e demanda pouco tempo para a execução das etapas de isolamento. |
The wild poinsettia (Euphorbia heterophylla L. Euphorbiaceae) is an annual weed species (Lorenzi et al., 2006), commonly found in crops grown in the South, Midwest and Southeast Brazil (Vargas et al., 1999). It presents the initial rapid development, large genetic variability (Trezzi et al., 2014) and high competitive capacity (Machado et al., 2015). In the study of the weeds, biotechnology has been increasingly employed, contributing to the investigation of the mechanisms and evolution of herbicide resistance (Tranel and Horvath, 2009), knowledge of genetic variability within and between species, in addition to serving as starting point for studies related to the management of these weeds.
The deoxyribonucleic acid (DNA) isolation process is the first step in the development of most molecular biology methodologies (Chen et al., 2010; Devi et al., 2013). Often, the reliability, viability and reproducibility of molecular genetic studies are still limited by the initial step of DNA isolation (Pereira et al., 2011). According to Chen et al. (2010) and Pereira et al. (2011) the best method for DNA isolation is one that optimizes the yield, minimizes the degradation, be reproducible and efficient in terms of labor, time and cost.
The commercial kits developed for DNA isolation are fast but high-cost, and generate large amounts of contaminant residues, while home isolation methods are cheap and much more effective, but generally the preparation process is slow (Mega and Revers, 2011). Most plant DNA isolation methods employ the method based on cationic detergent cetyltrimethylammonium bromide (CTAB), with modifications depending on the species (Kumari et al., 2012; Fehlberg et al., 2013). The major problems during the process of plant DNA isolation and purification are mainly due to the co-isolation of proteins, polysaccharides, secondary compounds including phenolic compounds (Japelaghi et al., 2011; Devi et al., 2013). Many species containing phenolic compounds involved in defense against herbivores (Boege and Marquis, 2006), such as E. heterophylla, in which latex production is abundant and can be observed both in the floral and vegetative parts (Cronquist, 1981). The latex is responsible for causing problems in good quality DNA isolation, due to contamination from phenolic compounds (Dhakshanamoorthy et al., 2013) released during cell lysis, which adhere to the DNA irreversibly inhibiting the amplifier by means PCR (polymerase chain reaction) (Couch and Fritz, 1990; Manoj et al., 2007; Dhakshanamoorthy et al., 2013).
The use of phenol, increase of the β-Mercaptoethanol concentration and use of polyvinylpyrrolidone (PVP) together with extraction buffer have been used in the isolation methods as alternatives to avoid the oxidative effect of polyphenols (Chaudhry et al., 1999; Deshmukh et al., 2007).
In the literature, a few works studying at the molecular level involving E. heterophylla employed DNA isolation methods based on cetyltrimethylammonium bromide (CTAB) in the presence or absence of phenol and β-Mercaptoethanol (BME) (Winkler et al., 2003; Winkler and Vidal, 2004), however, the quantity and quality of DNA from these studies have not been reported. Roso and Vidal (2010) by testing three methods of DNA isolation have found that the modified method of Murray and Thompson (1980) resulted in adequate quantity and quality of DNA isolated, but the interference in PCR was not evaluated.
Thus, to advance in the study of molecular genetics in E. heterophylla is of great importance to the development work aimed at optimizing the DNA isolation of the specie, seeking high yield DNA isolation combined with a high quality, which enable molecular studies subsequent as PCR reactions, sequencing and genotyping.
Given the above, this study aimed to determine the best low-cost method for the isolation of genomic DNA of the specie E. heterophylla.
The experimental design was completely randomized with four replications, where the treatments were constituted from 10 DNA isolation methods based on methods Doyle and Doyle (1987), with modifications Lefort and Douglas (1999) and Murray and Thompson (1980) with modifications Roso and Vidal (2010) (Table 1).
Young leaves of a single genotype of E. heterophylla were collected, wrapped in Falcon tubes 15 ml (Global Trade, Technology, Monte Alto, Brazil), and immediately frozen in liquid nitrogen preventing DNA degradation and contamination by polyphenols.
Table 1 – DNA isolation methods Doyle and Doyle (1987), with modifications Lefort and Douglas (1999) (0.2%BME) and Murray and Thompson (1980) with modifications Roso and Vidal (2010) (PHENOL).
Steps |
0.2%BME |
PHENOL |
1-Rupture of the cell walls |
Young leaves were ground to a fine powder in a mortar and pestle with liquid nitrogen |
Young leaves were ground to a fine powder in a mortar and pestle with liquid nitrogen |
2.1-Removal of lipids of cell membrane and exposure of the cell content |
Extraction Buffer A (TA) (100 ml): [1M] Tris-HCl, pH 8,0 (5 mL) [0,5M] EDTA (4 mL) [5M] NaCl (22 mL) CTAB (1 g) [10M] LiCl (4 mL) Tween 20 (0,5 g) PVP40 (2 g) Ultrapure H2O to complete. |
Extraction Buffer B (TB) (100 ml): [1M] Tris-HCl, pH 8,0 (5 mL) [0,5M] EDTA (4 mL) [5M] NaCl (22 mL) CTAB (2 g) Ultrapure H2O to complete. |
2.2-Removal of lipids of cell membrane and exposure of the cell content
|
In each tube of the 2 ml were added 0.2 g (~ 500 µL) of macerated leaf tissue and to this added 600 µL of extraction buffer (Vortex) (Table 2). The extraction buffer was preheated in a water bath at 60 °C for 15 minutes and immediately before use was added 0.2% BME. Then to each tube was added a small amount of PVPP (polyvinylpolypyrrolidone) using a 1 ml pipette tip with the tip cut in a bevel (Vortex). Samples were incubated at 60 °C for 25 minutes. |
In a 2 mL tube was added 0.2 g of macerated leaf tissue (~ 500 µL), 620 µL of extraction buffer B (Table 3) preheated in a water bath at 65 °C for 15 minutes, 6 µL/mL of proteinase K (20mg/ml) and 69 µL of SDS 20% (vortex). Samples were incubated at 65 °C for 1 h, stirring for 20 to 20 minutes. |
3-Removal of membrane proteins |
After removing the samples from the water bath is cooled at room temperature for 5 minutes. Then was added 600 µL of CIA (chloroform: isoamyl alcohol 24:1) and gently mixed by inverting tube for 4 minutes to form an emulsion. The samples were centrifuged at 10,000 x g. for 10 minutes at room temperature to separate the solid and aqueous phases. |
After removing the samples from the water bath were cooled at room temperature for 5 minutes. Was added 315 µL of phenol and immediately 315 µL of CIA (chloroform: isoamyl alcohol 24:1) and gently mixed by inverting up tube for 4 minutes to form an emulsion. Centrifuged at 10,000 x g. for 5 minutes at room temperature (25 oC) for separating the solid and aqueous phases. |
4-DNA precipitation |
The upper aqueous phase (500 µL) was transferred to a new 1.5 ml tube and added 250 µL 5M NaCl (Vortex) 750 µL of ice-cold isopropanol (-20 °C) (Vortex). Kept under refrigeration (4 to 6 °C) for 15-20 minutes. Then the samples were centrifuged at 10,000 x g. for 10 minutes at room temperature so that the DNA migrates to the bottom of the tube. |
Transferred the upper aqueous phase (500 µL) to a new 1.5 ml tube. Was added 333 µL of ice-cold isopropanol and stirred slowly. At this stage it was possible to see a cloud of DNA. Centrifuged at 10,000 x g. for 6 minutes to pellet the DNA. |
5-Wash DNA |
The supernatant was discarded and the pellet washed with 1000 μL 75% ethanol ice cream. The samples were subjected to quickly centrifugation (Spin) which contributes to the ethanol present in the wall of the tube migrates to the bottom, facilitating its removal with the aid of a micropipette. The DNA pellet was dried at 25 °C for 30 minutes. |
The supernatant was discarded and the pellet washed 2 times with 500 µL of cold 75% ethanol, and then centrifuged for 3 minutes and the supernatant was discarded. The DNA pellet was dried at 25 °C for 2 hours. |
6-Digestion of the RNA |
The pellet was resuspended in 50 µL of TE + RNAse (20μg/ml) incubated in a water bath at 37 °C for 15 minutes. Then, the DNA samples were stored at – 20 °C. |
The pellet was resuspended in 50 µL of TE + RNAse (20 µg/ml) and incubated in a water bath at 37 °C for 30 minutes. |
7-DNA Re-precipitation |
Steps not required. |
The DNA was re-precipitated in 5 µL sodium acetate (3M) and 100 µl of 96% ethanol, both ice (Vortex). maintained at -20 °C for 10 minutes. Centrifuged at 10,000 x g. for 5 minutes to pellet the DNA. The supernatant was discarded and the DNA pellet washed with 300 µl of cold 75% ethanol and dried at 25 °C for 30 minutes. The DNA was resuspended in 50 µl of TE + RNAse (20 µg/ml) incubated in a water bath at 37 °C for 10 minutes and then stored at -20 °C. |
The method of Doyle and Doyle (1987), with modifications from Lefort and Douglas (1999) (0.2% BME) was adapted for methods 1%BME, 2%BME, 3% BME, 4%BME and 5%BME which represent, respectively, 1, 2, 3, 4 and 5% β-mercaptoethanol added in 2.2 (Table 1).
Already, Murray method and Thompson (1980) with modifications from Roso and Vidal (2010) (PHENOL), was adapted for methods 3%BME+TA, 3%BME+TB and 3%BME+(TB+PVP40), that represent the addition of 3% β-mercaptoethanol in 2.2, replacing the phenol item 3 and, respectively, extraction buffer A, extraction buffer B and extraction buffer B plus polyvinylpyrrolidone (PVP40) in 2.1 (Table 1).
The DNA quantitation of the samples was performed in a digital spectrophotometer Shimadzu, UVmini-1240 model (SPLABOR, São Paulo, Brazil), assessing the optical density (OD) of the solution at a wavelength of 260 nm, calculating DNA concentration using the formula proposed by Sambrook and Russell (2001): [] DNA (ng µL-1) = OD260 x 50 x FD. Where, OD260 is the reading of the sample obtained at a wavelength of 260 NM, 50 is the conversion factor (where an OD260 equals 50 µg mL-1 double stranded DNA), and DF is the dilution factor used to conduct a reading in spectrophotometer. The DNA concentration is given in ng µL-1 equivalent to µg mL-1.
The nucleic acid by absorbing light at a wavelength of 260 NM and at 280 NM the proteins, by DNA ratio (OD260) and protein (OD280) as measured in a spectrophotometer verified the purity of the isolated DNA (Sambrook and Russell, 2001).
DNA integrity was checked by gel electrophoresis agorose 1% (Neuvitro Corporation, Washington, United States) (which were applied 1μL DNA dye nontoxic (AppliChem) + 4 μL DNA and 1 μL DNA dye non toxic + 3 μL molecular weight marker 1Kpb (Ludwig Biotec, Alvorada, Brazil) in TBE buffer (Tris-borate-EDTA) for 1 hour at 85 V. The gel was visualized under ultraviolet light on a transilluminator (Biosystems, Curitiba, Brazil) and the image was captured in a gel documentation system (VILBER, Eberhardzell, Germany).
Additionally, PCR was performed to verify the interference of possible contamination of the DNA using a pair of primers to amplify microsatellites. The PCR reaction was composed by: 7.5 ml ultrapure water, 5 μL of 10X buffer (100 mM Tris, 500 mM KCl, pH 8.3) (Ludwig Biotec, Alvorada, Brazil), 3 μL MgCl2 (50 mM) (Ludwig Biotec, Alvorada, Brazil), 2 μl of dNTP (10 mM) (Ludwig Biotec, Alvorada, Brazil), 2 μl of each EME19514 initiator forward 5’-GAAACTCCAGCACCAACAGA-3’ and Reverse 5’-ACACGGCCTCTTCTTCTATC-3’ (10 pmol μL-1), transferred from Manihot esculenta (Kunkeaw et al., 2011), 5 μL DNA (100 ng μL-1) and 0.5 μL Taq DNA polymerase (5 U μL-1) (Ludwig Biotec, Alvorada, Brazil), resulting in a final volume of 25 μl.
The PCR Microtubes (AXYGEN - Ciencor Scientific Ltda, São Paulo, Brazil) were placed in the thermocycler (DOGA limited, Ancara, Turkey) with the following steps: Step 1 (one cycle): 95 °C for 5 minutes; Step 2 (35 cycles): 95 °C for 1 minute; 53 °C for 1 minute; 72 °C for 1 minute; Step 3 (one cycle): 72 °C for 10 minutes.
The PCR products were subjected to agarose gel electrophoresis 2% (which were applied to the first well, 1 μL of DNA nontoxic colorant dye + 3 μL molecular weight marker 100 Pb (Norgen Biotek Corp., Toronto, Canada) and other wells, 2 μL DNA dye non toxic (AppliChem) + DNA 10 μL) in TBE buffer (Tris-borate-EDTA) for 1 hour at 85 V. The gel was visualized under ultraviolet light on a transilluminator the image was captured in a gel documentation system.
The results of the OD260/OD280 ratio and DNA concentration were analysed (analysis of variance) and the means were compared by test Scott-Knott hierarchical clustering algorithm (p=0.05) using the software Genes (Cruz, 2013).
The DNA isolation method 0.2%BME DNA showed a concentration of 7083 ng μL-1, statistically superior to the other methods (p=0.05) (Table 2). The quantity of DNA isolated by this method was much higher than the average obtained by Roso and Vidal with the modified method of Murray and Thompson (1980).
Thus, the concentration of 0.2% β-Mercaptoethanol was enough to avoid the oxidative effect of polyphenols, results also confirmed by Arriel et al. (2002) in DNA isolation sesame (Sesamum indicum L.) and this concentration suggested by Doyle and Doyle (1987) for DNA isolation from plants. Therefore, increased concentration of β-Mercaptoethanol above 0.2% for E. heterophylla DNA isolation is unnecessary, resulting only in an increase in insulation costs.
The DNA isolation methods 1% BME, 3% BME+(TB+PVP40) and 2%BME provided obtain 3695, 3245 and 2828 ng µL-1, respectively, the method being statistically lower than 0.2%BME and above the other methods (Table 2).
Smaller isolated DNA concentrations were obtained by the methods 3%BME+TA, 3%BME+TB, 4%BME, 5%BME, PHENOL and 3%BME not statistically different at 0.05 probability level (Table 2). These methods besides providing low amount of DNA isolated, using higher amounts of reagents or higher concentration increases the cost of isolation.
We found that increasing the concentration of β-Mercaptoethanol the method of Doyle and Doyle (1987) with modifications from Lefort and Douglas (1999) retained the DNA quality measured by the ratio OD260/OD280. Conversely, increasing β-Mercaptoethanol gave a decrease in the amount isolated DNA (Table 2), which corroborates with Borges et al. (2009) that found that the concentration of β-Mercaptoethanol increases the purity of the isolated DNA but reduces the amount.
The lower ratio OD260/OD280 to the isolated DNA was obtained with Method 3%BME+TB and the highest ratio obtained by PHENOL method, while the other methods presented intermediate values (Table 2).
Table 2 – Means of DNA concentration and OD260/OD280
ratio obtained by different isolation methods.
Methods |
[] DNA (ng µL-1) |
Ratio OD260/OD280 |
0.2%BME |
7083 a |
2.14 c |
1%BME |
3695 b |
2.14 c |
3%BME+(TB+PVP40) |
3245 b |
2.23 b |
2%BME |
2828 b |
2.13 c |
3%BME+TA |
2602 c |
2.19 b |
3%BME+TB |
2367 c |
2.05 d |
4%BME |
2096 c |
2.10 c |
5%BME |
2047 c |
2.12 c |
PHENOL |
1859 c |
2.30 a |
3%BME |
1634 c |
2.12 c |
CV (%) |
15.89 |
1.87 |
Treatments with averages followed by the same letter in the column are groups homogenous
statistically by Scott-Knott (P=0.05). []= Concentration. CV (%)= Coefficient of variation.
The OD260/OD280 ratio values in all methods were above 2, standing outside the purity range considered optimal for DNA is between 1.8 to 2.0 (Sambrook and Russell, 2001). According to Nelson and Cox (2004) values above the optimum range purity indicate DNA contamination of phenol and polyphenols, that are strongly absorbed wavelength of 280 NM.
The observation of higher values was optimal purity is due to the fact that the products used in the methods not being able to completely remove the phenolic compounds during the isolation justified by the high quantity of phenolic compounds produced by E. heterophylla (Cronquist, 1981).
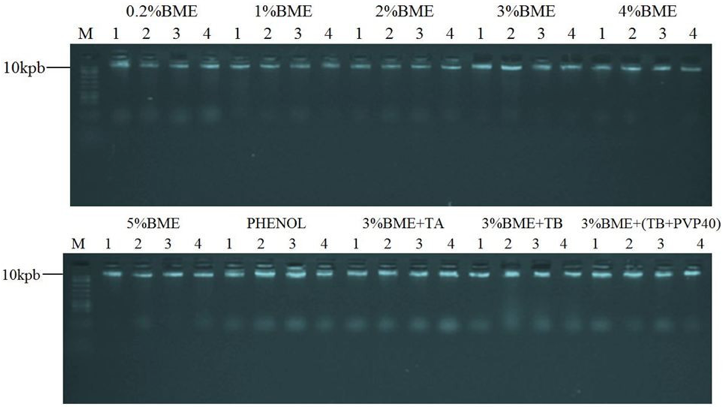
Problems with DNA smear in the gel and retention in the gel well was not observed for the tested methods (Figure 1) which indicate that the isolated DNA was not degraded by DNAses, no mechanical break during extraction with chloroform and the removal of polysaccharides was efficient. According to Romano and Brasileiro (1999) and Roso and Vidal (2010) are factors responsible for causing such problems.
Figure 1 – Agarose gel electrophoresis (1%) of genomic DNA of E. Heterophylla, obtained from the 10 different methods of isolation, with four replicates per method. 4 µL was used for each DNA sample, which remained migrating into the gel for 45 minutes at 85 V. Molecular weight marker 10 kbp (M).
DNAs isolated using the methods 0.2%BME, 1% BME, 2%BME, 3% BME, 4%BME and 5%BME adapted from Doyle and Doyle (1987), with Lefort and Douglas (1999) modifications showed no interference in PCR, thus demonstrating the ability DNA isolation quality (Figure 2).
Considering the adapted methods of Murray and Thompson (1980) with modifications Roso and Vidal (2010) no interference was observed in PCR for the DNA isolated by methods 3%BME+TA and 3%BME+(TB+PVP40) (Figure 2). However, the latter provided greater quantities of DNA isolated (Table 3). It is noteworthy that in both methods had the presence of PVP40. Since the DNA isolated by methods PHENOL and 3%BME+TB caused problems in PCR by the absence of bands (Figure 2).
The isolation of DNA with the phenol method resulted in contamination of the DNA by residual phenol or phenolic compounds which have not been efficiently removed by phenol, as there was no presence of PVP40 in extraction buffer, resulting in the absence of bands in the electrophoresis profile and the high ratio OD260/OD280 of 2.30. It is noteworthy that the DNA pellet with a characteristic brown aspect of contamination polyphenols was not observed in this method, indicating a greater likelihood that the contamination was caused only by phenol.
Even presenting ratio OD260/OD280 of 2.05 the DNA isolated from the method 3%BME+TB showed contamination by phenolic compounds, resulting in inhibition of PCR, possibly because there was no addition of PVP40 in extraction buffer. According Couch and Fritz (1990) e Devi et al. (2013) contamination from phenolic compounds prevents amplifications by PCR techniques, impairing the reproducibility of the analysis. These compounds oxidize DNA irreversibly (Kumari et al., 2012) contaminate the sample so that DNA begins to show brown colour (Jalal and Collin, 1977; Haq et al., 2009; Vaze et al., 2010), DNA aspect observed only in this method.
It was found through the method of isolation of 3%BME(TB+PVP40) the addition PVP40 to 2% in extraction buffer is sufficient to provide isolation of DNA quality for PCR, solving the problem of contamination from phenolic compounds observed in the method 3%BME+TB. This is possible because the addition of antioxidants such as PVP40 in extraction buffer binds to the phenolic compounds precipitating them, prevent their oxidative effect (Vaze et al., 2010).
Importantly, in all modifications in the methods of Doyle and Doyle (1987), with modifications Lefort and Douglas (1999) showed the presence of PVP40 2% in extraction buffer, a fact that likely contributed to no contamination from phenolic compounds.
Figure 2 – Profile electrophoretic agarose gel (2%) of genomic DNA amplification products of E. heterophylla, using SSR primer EME19514 transferred from Manihot esculenta, for the different methods of isolation with four replicates per method. molecular weight marker of 100 bp (M).

Among the isolation methods modified from Murray and Thompson (1980), with modifications Roso and Vidal (2010), method 3%BME+(TB+PVP40) showed no phenolics contamination problems, extracting DNA of sufficient quality. However, it was 54.18% in an amount less than 0.2%BME method, and requires much time for execution of the steps takes approximately 7-8 hours, nearly twice the time required for isolation by the method 0.2%BME which is approximately 4 hours.
The results obtained in this study it can be inferred that the 0.2%BME method adapted of Doyle and Doyle (1987), with modifications from Lefort and Douglas (1999) is the most suitable for DNA isolation of the species E. heterophylla. As were observed for polyphenols contamination problems, providing guarantee high quantity and quality of isolated DNA, as well require less time to perform the isolation steps.
The DNA isolation method 0.2%BME adapted from Doyle and Doyle (1987) with modifications from Lefort and Douglas (1999) is the most suitable for the species E. heterophylla. The addition of polyvinylpyrrolidone (PVP40) in extraction buffer addresses DNA contamination problems by phenolic compounds. The DNA isolation methods by 0.2%BME, 1%BME, 2%BME, 3%BME, 4%BME, 5%BME, 3%BME+TA and 3%BME+(TB+PVP40) enable molecular studies subsequent not inhibit the PCR reaction.
ARRIEL, N. H. C.; UNÊDA-TREVISOLI, S. H.; CAPELOTO, A.; MAURO, S. M. Z.; MAURO, A. O. (2002). Análise Comparativa de Quatro Protocolos de Extração de DNA Genômico, em Gergelim. Revista Brasileira de Oleaginosas e Fibrosas, v. 6, n. 2, p. 525-535.
BOEGE, K.; MARQUIS, R. (2006). Plant quality and predation risk mediated by plant ontogeny: consequences for herbivores and plants. Oikos, v. 115, n. 3, p. 559-572.
BORGES, A.; ROSA, M. S.; RECCHIA, G. H.; QUEIROZ-SILVA, J. R.; BRESSAN, E. A.; VEASEY, E. A. (2009). CTAB Methods for DNA Extraction of Sweetpotato for Microsatellite Analysis. Scientia Agricola, v. 66, n. 4, p. 529-534.
CHAUDHRY, B.; YASMEEN, A.; HUSNAIN, T.; RIAZUDDIN, S. (1999). Mini-scale Genomic DNA Extraction from Cotton. Plant Molecular Biology Reporter, v. 17, n. 1, p. 1-7.
CHEN, H.; RANGASAMY, M.; TAN, S. Y.; WANG, H.; SIEGFRIED, B. D. (2010). Evaluation of Five Methods for Total DNA Extraction from Western Corn Rootworm Beetles. PLOS One, v. 5, n. 8, p. e11963.
COUCH, J. A.; FRITZ, P. J. (1990). Isolation of DNA from plants high in polyphenolics. Plant Molecular Biology Reporter, v. 8, n. 1, p. 8-12.
CRONQUIST, A. (1981). An Integrated system of Classification of Flowering Plants. Columbia University Press, New York.
CRUZ, C. D. (2013). GENES - a software package for analysis in experimental statistics and quantitative genetics. Acta Scientiarum. Agronomy, v. 35, n. 3, p. 271-276.
DESHMUKH, V. P.; THAKARE, P. V.; CHAUDHARI, U. S.; GAWANDE, P. A. (2007). A simple method for isolation of genomic DNA from fresh and dry leaves of Terminalia arjuna (Roxb.) Wight and Argot. Electronic Journal of Biotechnology, v. 10, n. 3, p. 468-472.
DEVI, K. D.; PUNYARANI, K.; SINGH, N. S.; DEVI, H. S. (2013). An efficient protocol for total DNA extraction from the members of order Zingiberales- suitable for diverse PCR based downstream applications. SpringerPlus, v. 13, n. 2, p. 669.
DHAKSHANAMOORTHY, D.; SELVARAJ, R.; CHIDAMBARAM, A. (2013). Isolation of Genomic DNA from Five Species of Euphorbiaceae Without Liquid Nitrogen. Romanian Journal of Biology - Plant Biology, v. 58, n. 1, p. 3-8.
DOYLE, J. J.; DOYLE, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bulletin, v. 19, n. 1, p. 11-15.
FEHLBERG, S. D.; ALLEN, J. M.; CHURCH, K. (2013). A novel method of genomic DNA extraction for Cactaceae. Applications in Plant Sciences, v. 1, n. 3, p. 1200013.
HAQ, I.; KHAN, S.; DAHOT, M. U.; KOUSAR, N. (2009). Efficient plant genomic and viral DNA isolation from mature leaf midrib of banana (Musa spp). Journal of Plant Biochemistry and Biotechnology. v. 18, n. 2, p. 233-235.
JALAL, M. A. F.; COLLIN, H. A. (1977). Polyphenols of mature plant, seedling and tissue cultures of Theobroma cacao. Phytochemistry, v. 16, n. 9, p. 1377-1380.
JAPELAGHI, R. H.; HADDAD, R.; GAROOSI, G-A. (2011). Rapid and efficient isolation of high quality nucleic acids from plant tissues rich in polyphenols and polysaccharides. Molecular Biotechnology, v. 49, n. 2, p. 129-137.
KUMARI, V.; BANSAL, A.; AMINEDI, R.; TANEJA, D.; DAS, N. (2012). Simplified extraction of good quality genomic DNA from a variety of plant materials. African Journal of Biotechnology, v. 11, n. 24, p. 6420-6427.
KUNKEAW, S.; YOOCHA, T.; SRAPHET, S.; BOONCHANAWIWAT, A.; BOONSENG, O.; LIGHTFOOT, D. A.; TRIWITAYAKORN, K.; TANGPHATSORNRUANG, S. (2011). Construction of a genetic linkage map using simple sequence repeat markers from expressed sequence tags for cassava (Manihot esculenta Crantz). Molecular Breeding, v. 27, n. 1, p. 67-75.
LEFORT, F.; DOUGLAS, G. C. (1999). An efficient micro-method of DNA isolation from mature leaves of four hardwood tree species Acer, Fraxinus, Prunus and Quercus. Annals of Forest Science, v. 56, n. 3, p. 259-263.
LORENZI, H. (2006). Manual de identificação e controle de plantas daninhas: plantio direto e convencional. 6a ed. Nova Odessa – SP: Instituto Plantarum.
MACHADO, A. B.; TREZZI, M. M.; VIDAL, R. A.; PATEL, F.; CIESLIK, L. F.; DEBASTIANI, F. (2015). Rendimento de grãos de feijão e níveis de dano econômico sob dois períodos de competição com Euphorbia heterophylla. Planta Daninha, v. 33, n. 1, p. 41-48.
MANOJ, K.; TUSHAR, B.; SUSHAMA, C. (2007). Isolation and purification of genomic DNA from Black Plum (Eugenia jambolana Lam.) for analytical applications. International Journal of Biochemistry and Biotechnology, v. 3, n. 1, p. 49-55.
MEGA, N. O.; REVERS, L. F. (2011). Developing a rapid, efficient and low cost method for rapid DNA extraction from arthropods. Ciência Rural, v. 41, n. 9, p. 1563-1570.
MURRAY, M.; THOMPSON, W. F. (1980). Rapid isolation of high-molecular weight plant DNA. Nucleic Acids Res., v. 8, n. 19, p. 4321-4325.
NELSON, D. L.; COX, M. M. (2004). Lehninger, Principles of Biochemistry. 4a ed. New York: Worth Publishers.
PEREIRA, J. C.; CHAVES, R.; BASTOS, E.; LEITÃO, A.; GUEDES-PINTO, H. (2011). An Efficient Method for Genomic DNA Extraction from Different Molluscs Species. International Journal of Molecular Sciences, v. 12, n. 11, p. 8086-8095.
ROMANO, E.; BRASILEIRO, A. C. M. (1999). Extração de DNA de plantas. Biotecnologia, Ciência & Desenvolvimento, v. 2, n. 9, p. 40-43.
ROSO, A. C.; VIDAL, R. A. (2010). Determinação de Protocolo para Extração de DNA da Espécie Daninha Euphorbia heterophylla L. (EPHHL). In: Congresso Brasileiro da Ciência das Plantas Daninhas, 27., 2010, Ribeirão Preto. Anais. Ribeirão Preto: SBCPD.
SAMBROOK, J.; RUSSELL, D. W. (2001). Molecular Cloning: A Laboratory Manual. 4a ed. Cold New York: Spring Harbor Laboratory Press.
TRANEL, P. J.; HORVATH, D. P. (2009). Molecular Biology and Genomics: New Tools for Weed Science. BioScience, v. 59, n. 3, p. 207-215.
TREZZI, M. M.; MACHADO, A. B.; XAVIER, E. (2014). Soja: Impacto do leiteiro e estratégias de manejo. Cultivar, n. 183, p. 8-10.
VARGAS, L.; BORÉM, A.; SILVA, A. A. (1999). Técnicas de cruzamentos controlados em Euphorbia heterophylla L. Bragantia, v. 58, n. 1, p. 23-27.
VAZE, A.; NERKAR, G.; PAGARIYA, M.; DEVARUMATH, R. M.; THEERTHA PRASAD, D. (2010). Isolation and PCR Amplification of Genomic DNA from Dry Leaf Samples of Sugarcane. International Journal of Pharma and Bio Sciences, v. 1, n. 2, p. 1-6.
WINKLER, L. M.; VIDAL, R. A.; NETO, J. F. B. (2003). Caracterização genética de Euphorbia heterophylla resistente a herbicidas inibidores da acetolactato sintase. Pesquisa Agropecuária Brasileira, v. 38, n. 9, p. 1067-1072.
WINKLER, L. M.; VIDAL, R. A. (2004). Euphorbia heterophylla L. resistente aos herbicidas inibidores de acetolactato sintase: distribuição e genética de biótipos do estado do Paraná. Pesticidas: Revista de Ecotoxicologia e Meio Ambiente, v. 14, n. 1, p. 85-92.
1. Email: antoniopedro1991@hotmail.com